Welcome
Start exploring here! We hope this will be a useful resource to help you find the information you need about cerebral palsy and other childhood-onset disabilities. We want to help you to find answers to your questions – so please let us know what else you would like us to cover. Here we are presenting videos, summaries, research information and other resources.
Read on for more
Ways we can help
1
Family-friendly Books
Discover our collection of family-friendly books featuring accessible, research-based insights. These titles include the perspectives of families and individuals with lived experience, offering valuable guidance while supporting parents on their journey.
2
Free Chapters
We have selected a range of chapters from our books, offering helpful insights and practical tips. Our chapters summaries highlight key points. View the full chapter to explore each topic in more depth.
3
The Knowledge Bank
Explore the answers to your questions here. Find out more about conditions, treatments, interventions, and all aspects of care. Follow signposts to find more in-depth, evidence-based information from Mac Keith Press content, as well as other great sources of knowledge.
4
Plain Language Summaries
Plain language summaries are an effective way of communicating scientific research to a wider audience. By presenting the key findings and significance of a study in easy-to-understand language, the content becomes more accessible to more people. Here we present summaries of papers published in Developmental Medicine & Child Neurology (DMCN).
5
Helpful Videos
Here you will find a collection of short videos from authors and editors summarising their work. They cover Developmental Medicine & Child Neurology (DMCN) articles, Mac Keith Press books and e-learning. The aim of the videos is to help viewers get a clear understanding of why the research is important, how it was carried out, and real-world implications.
Gillette Children’s Healthcare Series
The goal of the Gillette Children’s Healthcare Series is to empower families through a greater understanding of their condition and therefore help optimize outcomes for children, adolescents and adults living with these childhood-acquired and largely lifelong conditions.

Spina Bifida
“Speaking from the perspective of someone living with spina bifida, I found this book does a phenomenal job of describing all the different ways the condition can affect a person. It also caters to different audiences—people with spina bifida, parents, and those who take care of people with the condition—so every reader can get something out of this book. The pictures do a really good job of showing how some people with spina bifida may function. The suggestions of how to exercise the muscles differently, too, are very helpful for someone who may struggle to find a solid workout routine. I learned some new things about spina bifida by reading this book that I will take with me as I continue my own journey of being an adult with the condition.”.”
Gracie Hadlich, Adult living with spina bifida
Click on ‘Buy Now’ to find out more!

Scoliosis: Congenital, Neuromuscular, Syndromic and other Nonidiopathic Types
“The team at Gillette Children’s has created an outstanding second book on scoliosis. This resource skillfully combines the latest clinical research with
personal stories, weaving together a rich tapestry of experiences that reflect the many diverse aspects of this condition. As a parent of a teenage daughter recently diagnosed with complex scoliosis, I understand the pressing need for accurate information and support. I often wished for a reference guide of this caliber during my search for information to help us navigate this journey. This uniquely practical guide offers clear language and step-by-step instructions, addressing the complexities of scoliosis throughout a person’s life. Having access to this information ultimately empowers families like ours to make informed decisions in collaboration with multidisciplinary teams, balancing clinical insights with the evolving needs of our children. I highly recommend this impactful book for families and professionals working in the field of scoliosis. Congratulations to everyone who contributed to this invaluable work, which brings hope and clarity to families living with scoliosis.”
Ann Marie Sutton, Parent of daughter with scoliosis, US
Click on ‘Buy Now’ to find out more!

Epilepsy
“Finally! A book that truly helps families understand epilepsy. As a mother of a special needs child with epilepsy, I found Epilepsy to be an invaluable resource. This book skillfully combines medical insights with heartfelt stories, addressing the real-life challenges families like ours face every day. … I wish I had this book at the beginning of my journey. It’s a must-read for any family navigating life with epilepsy.”
Colleen Peterson, parent of son with Wolf-Hirshhorn and Lennox-Gastaut syndromes, USA
Click on ‘Buy Now’ to find out more!

Spastic Diplegia
“This is an indispensable guidebook for navigating spastic diplegia, as it is written for families with the condition. It imparts a deep understanding of the medical science and treatment pathways, supported by comprehensive evidence-based references and information resources. The author’s generous sharing of her and her son’s journey, and those of many others, provides valuable sign-posting, hope, and inspiration for the reader.”
Jean and John Glynn, parents of a son with spastic diplegia, Ireland
Click on ‘Buy Now’ to find out more!

Spastic Hemiplegia
“As someone with right spastic hemiplegia, I was amazed by how much of the information was relevant to my memories of childhood therapy appointments and doctor visits. I deeply enjoyed reading the testimonies of those with CP, and I felt recognized inside their stories. From small things like official medical terminology to detailed explanations on why I was receiving certain treatments as a child, this book helped me not only recontextualize my own experiences but also prepared me to be a better medical advocate for myself moving into adulthood.”
Emmalynne Shumard, Student; Adult with spastic hemiplegia, US
Click on ‘Buy Now’ to find out more!

Spastic Quadriplegia
“This book presents clear and concise explanations and eliminates the confusion caused by misinformation online. It has helped me realize that we are not alone; there are other families just like ours experiencing the same highs and lows, joys and sorrows. It will provide comfort and hope to families striving to adjust to a new and oftentimes difficult diagnosis.”
Kristen Stier, Mother of a young adult with spastic quadriplegia, US
Click on ‘Buy Now’ to find out more!

Craniosynostosis
“This is an excellent and informative book that is both clear and factual. It serves as a valuable resource for families, providing them with essential knowledge about craniosynostosis and empowering them to engage confidently with medical and health professionals. The personal stories included offer hope and reassurance, reminding families that they are not alone in their journey. This is the book I wish I had when my son was diagnosed with sagittal craniosynostosis in 2019.”
Elaine L. Kinsella, Parent; Chartered Psychologist and Associate Professor in Psychology, University of Limerick, Ireland
Click on ‘Buy Now’ to find out more!

Idiopathic Scoliosis
“This book is great for anyone on this journey! Our daughter was diagnosed with juvenile idiopathic scoliosis right before she started kindergarten, and we were so worried and overwhelmed, and had so many questions. We wish we had this book during that time as it answers so many questions. We still have many questions and this book helps us pave a path today and for the future.”
Amber Marlatt, Parent of daughter with juvenile idiopathic scoliosis, US
Click on ‘Buy Now’ to find out more!
Free chapter downloads

Feeding and Nutritional Management Strategies
Nutrition and Neurodisability highlights managing feeding and nutrition in children with neurological impairments. Chapter 9 highlights how a multidisciplinary team approach can improve feeding safety and efficiency, addressing issues like dysphagia, poor nutrition, and gastrointestinal problems. Interventions include oral nutrition support, tube feeding, and caregiver training.

Personal experiences from a young person with visual impairment, Holly Tuke
This chapter is a personal account by Holly Tuke, a woman with retinopathy of prematurity, which caused blindness. Despite challenges with accessibility and independence, Holly excelled academically with support and assistive technology. Now a university graduate, she works in the charity sector and runs a successful blog, advocating for disability awareness.

Cerebral Palsy: Through the Eyes of Parents
Parents raising children with complex cerebral palsy (CP) face emotional, practical, and social challenges, including balancing caregiving with personal needs. Support from clinicians, fostering the child’s independence, and improving quality of life through recreational activities are essential. Long-term care planning and focusing on the child’s interests are also important.

Life with and for a Person with Down Syndrome
Families of children diagnosed with Down syndrome often face concerns about health, development, and support. It’s essential to deliver the diagnosis with care and provide accurate, up-to-date information. Early health assessments, addressing feeding challenges, and fostering development are key to helping children thrive, supported by love, encouragement, and healthcare guidance.

What is Cerebral Palsy?
‘Cerebral palsy (CP) describes a group of permanent disorders of the development
of movement and posture, causing activity limitation, that are attributed
to non-progressive disturbances that occurred in the developing fetal or infant
brain. The motor disorders of cerebral palsy are often accompanied by disturbances
of sensation, perception, cognition, communication, and behaviour,
by epilepsy, and by secondary musculoskeletal problems.’ This is the official (2007) definition – read the full chapter to find out much more.

How to Promote a Physically Active Lifestyle Across the Lifespan
The Knowledge Bank
Explore the answers to your questions here. Find out more about conditions, treatments, interventions, and all aspects of care. Follow signposts to find more in-depth, evidence-based information from Mac Keith Press content, as well as other great sources of knowledge.
What are the different types of craniosynostosis?
There are multiple classifications of CS. Generally, it is classified based on the cause of the condition, the number of sutures involved, and the sutures that fused prematurely, resulting in a specific head shape.
What are the different types of craniosynostosis?
There are multiple classifications of CS. Generally, it is classified based on the cause of the condition, the number of sutures involved, and the sutures that fused prematurely, resulting in a specific head shape.
This answer is adapted from the Gillette Children’s Healthcare Series book on Craniosynostosis.
Classification by cause
- Nonsyndromic CS: CS that is not associated with a syndrome but is instead its own medical condition that has no known cause. Nonsyndromic CS accounts for about 85% of cases and most are single-suture fusions.
- Syndromic CS: Occurs as part of a genetic syndrome (e.g., Apert, Crouzon, Pfeiffer syndromes). Syndromic CS accounts for about 15% of cases, usually involves multiple sutures and is more complex to treat.
Classification by the number of Sutures Involved
- Single-suture CS: Only one suture fuses prematurely.
- Multisuture CS: More than one suture is fused; commonly syndromic and more severe.
Classification by which suture fused (head shape patterns)
- Sagittal CS (Scaphocephaly): Sagittal suture fuses causing a long, narrow, boat-shaped head.
- Metopic CS (Trigonocephaly): Metopic suture fuses. Creating a triangular forehead, closely spaced eyes.
- Unicoronal CS (Anterior Plagiocephaly): One coronal suture fuses making the forehead asymmetrical with one side flat (eye and nose may shift).
- Bicoronal CS (Brachycephaly): Both coronal sutures fuse creating a short, wide head with tall/flat forehead.
- Lambdoid CS (Posterior Plagiocephaly): One lambdoid suture fuses leading to flattening at the back of the head on one side.
How is craniosynostosis diagnosed?
Craniosynostosis may be suspected when an infant presents with an atypical head shape, usually noticed shortly after birth by parents, family members, or medical professionals familiar with the condition.
How is craniosynostosis diagnosed?
This answer is adapted from section 1.5 of the Gillette Children’s Healthcare Series book on Craniosynostosis.
Craniosynostosis may be suspected when an infant presents with an atypical head shape, usually noticed shortly after birth by parents, family members, or medical professionals familiar with the condition. In rare cases, particularly with syndromic forms, it may be identified before birth by ultrasound, although prenatal diagnosis is uncommon. When craniosynostosis is suspected, the child is referred to a pediatric craniofacial surgeon within a specialized hospital, where diagnosis and management are carried out by a multidisciplinary craniofacial team, often in partnership with a neurosurgeon.
Diagnosis begins with a detailed medical and family history, including information about pregnancy, maternal health or medication use, family history of craniosynostosis, and any feeding or breathing difficulties after birth, which are more typical in syndromic cases. Head growth patterns are also reviewed to identify growth that is unusually fast or slow. This is followed by a thorough physical examination. Clinicians assess facial symmetry, palpate sutures for ridging or flatness, and examine the fontanels for abnormal shape or persistent bulging or sinking. Eye movements and reflexes are checked, with referral to an ophthalmologist recommended for all infants with craniosynostosis. Head circumference is measured and compared with World Health Organization growth charts, though normal circumference does not rule out the condition due to compensatory skull growth. A more accurate indicator is the cranial index, which compares the width and length of the skull. Neurological exams may also be performed if there are concerns about reflexes, movement, or sensation.
If clinical suspicion remains high, imaging is ordered to confirm the diagnosis. Computed tomography (CT) is the most commonly used and reliable imaging technique, providing detailed images of fused sutures and allowing for three-dimensional reconstructions. X-rays can also visualize bone structures but are less comprehensive. Magnetic resonance imaging (MRI) may be used to assess brain tissue and development, offering the advantage of no radiation exposure, though it often requires sedation and is less effective for bone evaluation. Ultrasound, which uses sound waves instead of radiation, may also be used in certain cases to examine sutures.
What does “syndromic” craniosynostosis mean and how is this different from “nonsyndromic” craniosynostosis?
Syndromic craniosynostosis is a form of craniosynostosis that occurs as part of a broader syndrome (a condition defined by a recognizable group of characteristics that consistently appear together). In contrast, nonsyndromic craniosynostosis is not linked to any underlying syndrome and occurs as a standalone condition with no identifiable cause.
What does “syndromic” craniosynostosis mean and how is this different from “nonsyndromic” craniosynostosis?
This answer is adapted from chapter 3 of the Gillette Children’s Healthcare Series book on Craniosynostosis.
Syndromic craniosynostosis is a form of craniosynostosis that occurs as part of a broader syndrome (a condition defined by a recognizable group of characteristics that consistently appear together). It is usually caused by genetic changes and accounts for about 15 percent of all craniosynostosis cases, making it less common than nonsyndromic craniosynostosis.
Because syndromes are diagnosed by recognizing patterns of features rather than a single test (except in cases with a known genetic mutation), individuals with syndromic craniosynostosis may show several, but not necessarily all, traits of the associated syndrome. Importantly, nearly 200 different syndromes have been linked to craniosynostosis, though not all people with those syndromes will necessarily develop craniosynostosis.
In contrast, nonsyndromic craniosynostosis is not linked to any underlying syndrome and occurs as a standalone condition with no identifiable cause. It is the most common type, representing about 85 percent of cases.
What are the common syndromes associated with craniosynostosis?
There are five most prevalent craniosynostosis syndromes: Apert syndrome; Crouzon syndrome; Pfeiffer syndrome; Muenke syndrome; Saethre-Chotzen syndrome.
What are the common syndromes associated with craniosynostosis?
This answer is adapted from section 3.2 of the Gillette Children’s Healthcare Series book on Craniosynostosis.
The five most prevalent CS syndromes are:
- Apert syndrome: Marked by distinctive skull and face shape, and syndactyly (webbed or fused fingers and toes), often with breathing, feeding, and brain development challenges.
- Crouzon syndrome: Characterized by bulging eyes, midface underdevelopment, and hearing loss; severity varies widely, and some individuals are unaware until genetic testing.
- Pfeiffer syndrome: Features high forehead, abnormal hand/foot bones, and possible organ issues; ranges from mild (type 1) to severe (types 2 and 3 with cloverleaf skull, airway obstruction, and cognitive delays).
- Muenke syndrome: Involves wide-set eyes and flattened cheekbones, usually without limb abnormalities; about 20% of cases occur without CS.
- Saethre-Chotzen syndrome: Presents with variable skull, face, hand, and foot differences, sometimes very mild; some individuals remain unaware until genetic testing.
Submit a question
Let us know what other questions you have. Are there specific topics you would you like us to cover?
Contact us now using the form.
Plain Language Summaries
Plain language summaries are an effective way of communicating scientific research to a wider audience. By presenting the key findings and significance of a study in easy-to-understand language, the content becomes more accessible to individuals with disabilities, parents, caregivers, and others. Here we present summaries of papers published in Developmental Medicine & Child Neurology (DMCN).

Sleep problems in children with cerebral palsy and their parents
The study found that sleep problems are common in children with cerebral palsy (CP) aged 0 to 11 years. The most common problems reported by parents were: daytime fatigue, difficulty falling asleep, and early-morning waking. Children with CP were also more likely to have sleep problems than typically developing children, and children with CP who cannot walk were more severely affected by sleep problems than children with CP who can walk.

Quality of life in caregivers of a child with a developmental and epileptic encephalopathy
Developmental and epileptic encephalopathies (DEEs) are severe forms of epilepsy that usually start during infancy. There are many types of DEE, making each specific diagnosis extremely rare. Children with a DEE typically have seizures that are hard to treat and that impact their development and learning. Other symptoms may include feeding difficulties, movement problems, and features on the autism spectrum.

Experiences of children and adolescents with attention-deficit/hyperactivity disorder taking methylphenidate
Understanding the experiences of adolescents diagnosed with attention-deficit/hyperactivity disorder (ADHD) and taking ritalin and other stimulant medication is crucial in order to improve medical counselling to them. A central theme in these experiences is adolescents’ self-esteem and their sense of control on their body and life.

Epilepsy and cannabis: so near, yet so far
In 2018, the UK government changed the law so that cannabis-based medicines could legally be prescribed. Since then, very few prescriptions have been issued. Why is this?
Some children with epilepsy have seizures that respond very poorly to standard medications and their quality of life suffers. CBMPs may have an important role in helping those children. There are many forms of CBMPs and one medicine, cannabidiol, now has a license for some rare types of epilepsy.
Child-led goal setting and evaluation tools for children with a disability: A scoping review
Children with disabilities and delays benefit from being involved in setting and evaluating intervention goals. When goals hold personal value for children, they can feel more motivated to work towards them, which can improve their intervention outcomes. However, in current practice, parents or therapists are most often the primary decision-makers about intervention priorities. Which practices support allied health professionals to involve children with disabilities in goal setting and evaluation?
‘Power in Mobility’: Parent and therapist perspectives of the experiences of children learning to use powered mobility
This study focuses on the importance of mobility for children with mobility impairments and the impact of using powered mobility devices on their development and participation. To gather data, interviews with parents and therapists of children who had recently started using powered mobility devices were analyzed to identify common themes and gain insights into the experiences and perceptions of parents and therapists.
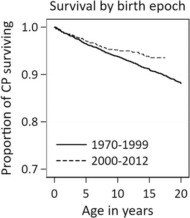
Survival of individuals with cerebral palsy: A Victorian longitudinal cohort study spanning four decades
The aim of this research was to provide an updated description of the death rates, trends in death rates over time, and predictors of deaths of persons with cerebral palsy (CP) who were born in the Australian state of Victoria between 1970 and 2012. The authors found that improvements in survival for those born in the 2000s was likely mainly related to a proportional reduction in complex CP, a finding that is supported by other studies.
Letting Tourette’s be: The importance of understanding lived experience in research and the clinic
Tourette syndrome is a neurodevelopmental condition characterized by involuntary movements and sounds that are known as tics. Historically, the focus of biomedical and clinical research and treatment has been on reducing these tics, viewing them primarily as symptoms of a neurological disorder. However, in this article we argue that this approach is too narrow as it does not adequately consider the lived experiences of Tourettic individuals.

UK research priority setting for childhood neurological conditions
In this project, the researchers wanted to find the most important unanswered questions about treatments, or therapies for children and young people with childhood neurological conditions such as epilepsies, cerebral palsy, and many rare conditions. This is called a Priority Setting Partnership. Priority Setting Partnerships aim to help patients, carers, and health professionals work together to agree research priorities. After two rounds of surveys, the top 10 priorities were identified.

Environment-based approaches to improve participation of young people with physical disabilities during COVID-19
Personalized interventions to enhance participation in meaningful activities in everyday environments are recommended for young people with physical disabilities. Pathways and Resources for Engagement and Participation (PREP) is one such intervention, focusing on changing the environment (e.g. inaccessibility, limited social support, lack of availability of programs) and coaching young people/parents and community members on removing environmental barriers.

Magic-themed motor training for daily bimanual task performance in children with unilateral spastic cerebral palsy: A systematic review and meta-analysis
Unilateral spastic cerebral palsy (CP) is a disorder of motor and postural development caused by early brain injury. This impairment poses significant challenges for daily physical tasks such as getting dressed, taking a shower, cutting food, etc. The authors of this study undertook a systematic literature review to discover what research has taken place on the effectiveness of magic-themed interventions in improving task performance in both hands in children with unilateral spastic CP.
Helpful videos
Here you will find a collection of short videos from authors and editors summarising their work. They cover Developmental Medicine & Child Neurology (DMCN) articles, Mac Keith Press books and e-learning. The aim of the videos is to help viewers get a clear understanding of why the research is important, how it was carried out, and real-world implications.

Contact us
Please feel free to contact us if you have any feedback or suggestions: